Cell-free Chemoenzymatic Starch Synthesis From Carbon Dioxide
From carbon dioxide to starch: no plants required

Authors: Tao Cai, Hongbing Sun, Jing Qiao, Leilei Zhu, Xfan Zhangjie, Zhangzijing Tang, Xinlei Wei, Jiangang Yang, Qianqian Yuan, Wangyin Wang, Xue Yang, Huanyu Chu, Qian Wang, Chun You, Xhongwu Ma H, Yuanxia Sun, Yin Li, Can Li, Huifeng Jiang, Qinhong Wang H, and Yanhe Ma.
Many plants turn glucose from photosynthesis into polymers that form insoluble starch granules ideal for long-term energy storage in roots and seeds. Cai et al. developed a hybrid system in which carbon dioxide is reduced to methanol by an inorganic catalyst and then converted by enzymes first to three and six carbon sugar units and then to polymeric starch. This artificial starch anabolic pathway relies on engineered recombinant enzymes from many different source organisms and can be tuned to produce amylose or amylopectin at excellent rates and efficiencies relative to other synthetic carbon fixation systems—and, depending on the metric used, even to field crops. —MAF
Abstract
Starches, a storage form of carbohydrates, are a major source of calories in the human diet and a primary feedstock for bioindustry. We report a chemical-biochemical hybrid pathway for starch synthesis from carbon dioxide (CO2) and hydrogen in a cell-free system. The artificial starch anabolic pathway (ASAP), consisting of 11 core reactions, was drafted by computational pathway design, established through modular assembly and substitution, and optimized by protein engineering of three bottleneck-associated enzymes. In a chemoenzymatic system with spatial and temporal segregation, ASAP, driven by hydrogen, converts CO2 to starch at a rate of 22 nanomoles of CO2 per minute per milligram of total catalyst, an ~8.5-fold higher rate than starch synthesis in maize. This approach opens the way toward future chemo-biohybrid starch synthesis from CO2.
Starch is a main caloric component of food and animal feed, as well as an important industrial feedstock (1, 2). Amylose and amylopectin polymers in starch granules consist of chains of glucosyl residues linearly linked by α-1,4-glycosidic bonds, interspersed by branching points of α-1,6-glycosidic bonds in the case of amylopectin (3). Starch synthesis in green plants involves about 60 steps and complex regulation (4, 5). Although many efforts have been made to improve the production of starch in plants (6–8), the inefficiency of photosynthesis and the complexity of starch biosynthesis are obstacles (9). By contrast, advances in synthetic biology have enabled the design and construction of synthetic systems for more efficient CO2 fixation (10–14) and chemical production (15, 16). Inspired by the central principles of photosynthesis, extraordinary chemical catalysts have been developed to provide electrons (17) or hydrogen (18) more efficiently from solar energy and water for reducing CO2 into simple chemicals (19, 20). In this study, we used a chemical CO2 reduction catalyst that produces reduced one-carbon (C1) units as an input to a chemoenzymatic pathway for cell-free starch synthesis.
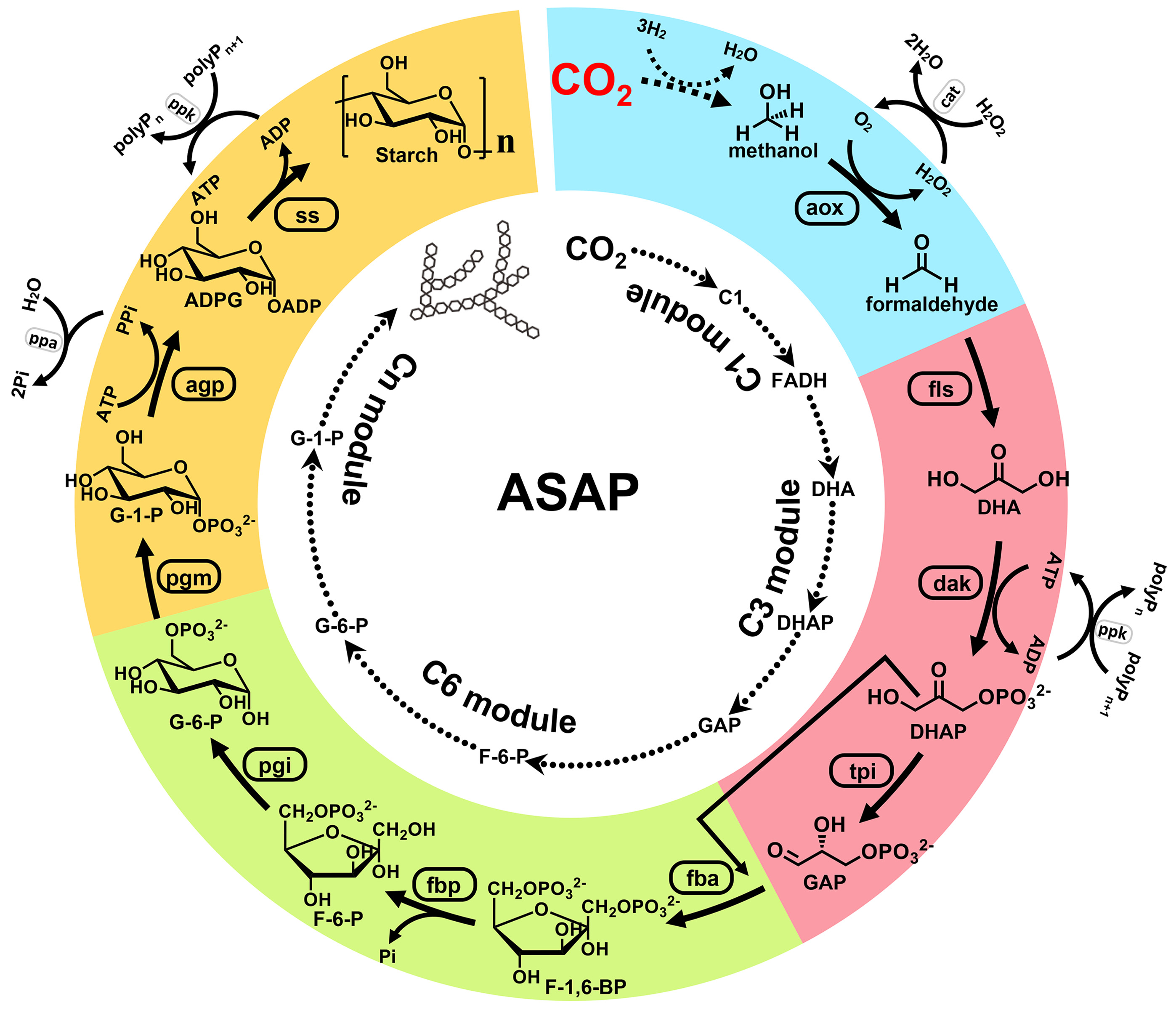
To build this hybrid pathway, we first chose formic acid and methanol to serve as the candidate intermediates to bridge possible chemical catalysts and biological enzymes. We exploited formolase (fls) to design and construct the enzymatic part of the starch synthesis pathway from the candidate C1 intermediates (21). On the basis of a main set of 6568 reactions from the MetaCyc database (22) and ATLAS database (23) and two combinatorial sets of 15 formate and 8 methanol utilization reactions, we drafted two concise starch synthesis pathways from either formic acid or methanol by using the combination of combinatorial algorithm and parsimonious flux balance analysis (comb-FBA) (24) and the COBRApy toolbox in Python (25) (fig. S1A and supplementary text). Starch could be synthesized, in principle, through only nine core reactions from CO2 with formic acid or methanol as the C1 bridging intermediate (Fig. 1, inner circle).

Fig. 1. Design and modular assembly of an artificial starch anabolic pathway.Inner circle: schematic of the artificial starch pathway drafted by computational pathway design with divided modules. C1 here indicates formic acid and methanol. Outer circle: schematic of artificial starch anabolic pathway (ASAP) 1.0, with individual modules colored. Auxiliary enzymes and chemicals are indicated. ADPG, ADP glucose; aox, alcohol oxidase; FADH, formaldehyde; F-1,6-BP, D-fructose-1,6-bisphosphate; F-6-P, D-fructose-6-phosphate; GAP, D-glyceraldehyde 3-phosphate; pgi, phosphoglucose isomerase; polyP, polyphosphate; pgm, phosphoglucomutase; ppa, pyrophosphatase; ppk, polyphosphate kinase; ss, starch synthase; tpi, triose-phosphate isomerase.
In contrast to natural pathways that have evolved functionality and compatibility over hundreds of millions of years of selection, computationally designed pathways are often hindered by unpredictable and undesired interactions between enzymes from disparate biochemical contexts (26). To overcome these problems, we pursued a strategy of modular assembly and substitution. Two starch synthesis pathways were divided into more manageable modules (fig. S1A), including a C1 module (for formaldehyde production), a C3 module (for D-glyceraldehyde 3-phosphate production), a C6 module (for D-glucose-6-phospate production), and a Cn module (for starch synthesis). According to known enzymes in databases, five modules were initially constructed (C1a/C1b, C3a, C6a, and Cna). Although the C1a, C1b, and C3a modules displayed function individually (figs. S2, A, B, and G, and S3), the assembly of C1a or C1b with C3a did not result in detectable C3 compounds from formic acid or methanol (fig. S6A). We speculated that the marginal formaldehyde production from energy-efficient but thermodynamically unfavorable C1 modules may not be able to supply material for the key reaction of fls in the C3a module (supplementary text). We thus constructed the alternative C1 module with thermodynamically more favorable reaction cascades (fig. S2, C to E, C1c to e) (21, 27). The most thermodynamically favorable C1e module was successfully assembled with the C3a module and achieved a substantially higher yield of C3 compounds from methanol (fig. S6A).
Assembling C1e + C3a with the C6a module (fig. S4A) produced negligible amounts of the target glucose-6-phosphate (G-6-P) (fig. S6B). We found the carbon flux was kinetically trapped at triose phosphates (fig. S8A) because of the unbalanced activity between dihydroxyacetone kinase (dak) in module C3a and fructose-6-phosphate aldolase (fsa) in module C6a (fig. S8, B and D). Furthermore, glycolaldehyde, which is the by-product of fls-catalyzed reaction in C3a, competitively inhibited the function of fsa (fig. S8, C and D, and supplementary text for more details of the incompatibilities between the C3a and C6a modules). Two alternative modules were constructed on the basis of different classes of aldolase (fig. S4, B and C, C6b and c). However, the extremely low activity of thermophilic fructose-1,6-bisphosphate aldolase/phosphatase at ambient temperature impeded the performance of C6c (table S1 and fig. S4E). For assembly of (C1e + C3a) + C6b, the function of Escherichia coli fructose-bisphosphatase (fbp) of module C6b was inhibited by adenosine 5′-triphosphate (ATP) and adenosine 5′-diphosphate (ADP), the essential cofactors of dak in the C3a module (fig. S9, A and B, and supplementary text). By coupling an ATP regeneration system with (C1e + C3a) + C6b [designated as (C1e + C3a) + C6b*], in which ATP was regenerated from ADP by consuming polyphosphate via polyphosphate kinase (table S1), we reduced ATP and ADP to a tolerable level and successfully produced G-6-P from methanol (figs. S9B and S6B).
Assembling C1e + C3a + C6b* with the Cna module (fig. S5A) failed to produce detectable amylose starch from methanol (fig. S6C). We observed that amylose synthesis via α-glucan phosphorylase of Cna was severely inhibited by the high inorganic phosphate (Pi)/α-D-glucose-1-phosphate (G-1-P) ratio, which could be formed from the upper part of the assembly (fig. S10, A and B, and supplementary text). Alternatively, we constructed an ATP-dependent Cnb module (fig. S5B), which is resistant to a high Pi/G-1-P ratio (fig. S10C). The assembly of (C1e + C3a + C6b*) + Cnb enabled 30 mg liter−1 amylose starch production from 20 mM methanol (Table 1 and fig. S6C).

Table 1. Comparison of ASAP iterations with other natural and synthetic pathways.The average growth period of maize was assumed as 120 days. These numbers may vary depending on species, geographic location, and cultivation practices. The average molar weight of carbon unit in starch of maize was assumed as 27 g mol−1. We note that starch synthesis in maize is considerably more complex than in our in vitro chemoenzymatic ASAP. For ASAP iterations, values are means, and error bars indicate SD (n = 3 replicates). NADPH, reduced form of nicotinamide adenine dinucleotide.
*Starch synthesis rate was calculated for the indicated substrates and shown as a nanomole of carbon converted to product per minute per milligram of total proteins.
†The rate was calculated by using total amount of both catalyst and proteins (see supplementary materials).
‡The end product of CETCH is glyoxylate. Rate of CETCH was recalculated with 3.1 mg ml−1 of total proteins (13).
§Calculation mainly based on reported data that starch is 26.1% of total biomass and total proteins, excluding storage protein in grain, are 2.17% of total biomass (40).
With the assistance of computational pathway design and through assembling and substituting 11 modules constructed from a pool of 62 enzymes from 31 organisms (table S1), we established the artificial starch anabolic pathway (ASAP) 1.0 with 10 enzymatic reactions starting with methanol (Fig. 1, outer circle). The main intermediates and target product of ASAP 1.0 were detected by an isotopic 13C-labeling experiment (fig. S11, A and C), validating its full function for starch synthesis from methanol.
After establishing ASAP 1.0, we sought to optimize this pathway by resolving potential bottlenecks. First, because of its low kinetic activity, the enzyme fls accounted for ~86% of the total protein dosage in ASAP 1.0 to sustain the metabolic flux and maintain toxic formaldehyde at a very low level (28, 29). Directed evolution increased the fls catalytic activity, yielding the variant fls-M3 (flsI28L/T90L/N283H), which showed an activity improvement of 4.7-fold toward 5 mM formaldehyde and a preference of dihydroxyacetone (DHA) as the main product (fig. S12 and supplementary text).
Even though they were maintained at a low level of 1 mM with the assistance of the regeneration system, ATP and ADP may still partially inhibit the function of E. coli fbp (Fig. 2, B and C), which is reported to be allosterically inhibited by adenosine 5′-monophosphate (AMP) (30). We found that the variant fbp-AR (fbpK104Q/R132I), which contains two mutations in the AMP allosteric site (31), alleviated ADP inhibition (Fig. 2, B and C) and substantially improved G-6-P production from DHA (Fig. 2D). Analysis of the inhibition pattern of the three kinds of nucleotide on fbp and fbp-AR indicated that ATP or ADP was the determinant for inhibition in the system (table S5 and supplementary text). By integrating fbp-AR with a reported variant resistant to G-6-P (31), a combined variant fbp-AGR (fbpK104Q/R132I/Y210F/K218Q) enabled a further improvement (Fig. 2D and supplementary text).
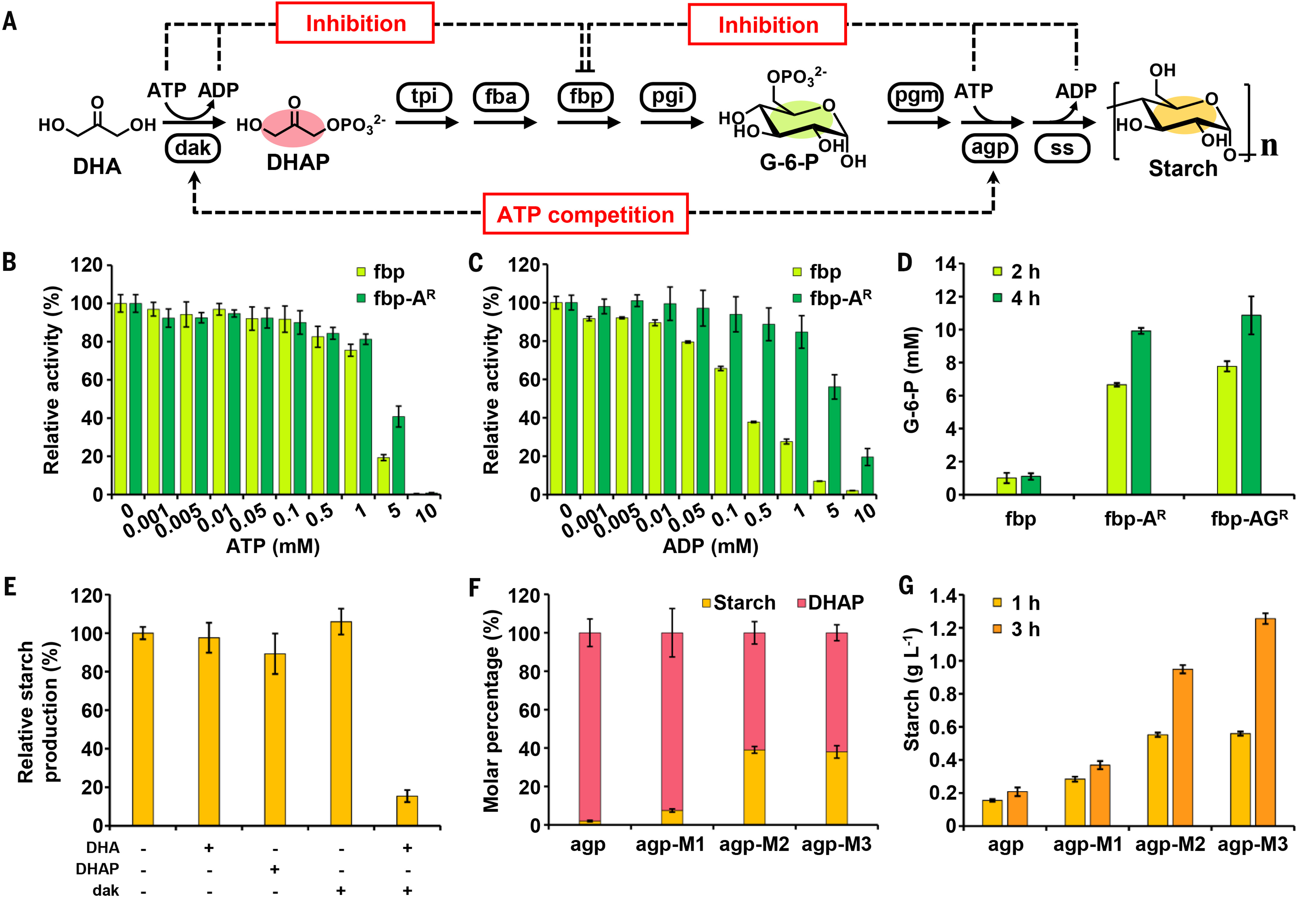
Fig. 2. Resolving main bottlenecks in ASAP.(A) Partial ASAP pathway from DHA to starch with bottlenecks indicated and key intermediates and product colored. (B and C) Inhibitory effect of ATP and ADP on fbp and fbp-AR. (D) G-6-P production from 25 mM DHA via pathway with different fbp variants. (E) Inhibition by components from C3a model on the Cnb model. DHA, DHAP, and dak were present at concentrations of 25 mM, 10 mM, and 0.2 mg ml−1, respectively. (F) Proportion of DHAP and starch (in glucose) produced from 25 mM DHA and 10 mM G-6-P in a competitive system, which includes dak and the Cnb module, with different agp variants. (G) Starch (in glucose) production from 25 mM DHA via partial ASAP as depicted in Fig. 2A. For (B) to (G), values are means, and error bars indicate SD (n = 3 replicates).
ATP competition between dak and ADP-glucose pyrophosphorylase (agp) was considered, as an increase in substrate DHA and its kinase dak resulted in an aberrantly lower starch production during the first 4 hours (Fig. 2A and fig. S10D). We confirmed that the coexistence of DHA and dak severely inhibited starch synthesis via Cnb (Fig. 2E) and output DHA phosphate (DHAP) as the dominant product over starch (Fig. 2F, first column), which validates that dak competitively consumed most of the ATP. Instead of reducing the dosage of dak, we tried to enhance the capacity of agp. Three high-activity agp variants were created in accordance with reported amino acid substitutions (32, 33), and these variants displayed enhanced competition against dak (Fig. 2F). The best variant, agp-M3, successfully increased starch synthesis from DHA by approximately sixfold (Fig. 2G).
Using these three engineered enzymes (fls-M3, fbp-AGR, and agp-M3), we constructed ASAP 2.0, which produced ~230 mg liter−1 amylose starch in 10 hours from 20 mM methanol (Table 1). Compared with that of ASAP 1.0, the starch productivity of ASAP 2.0 was improved 7.6-fold. On the basis of 13C-labeling liquid chromatography–mass spectrometry (LC-MS) analysis, ASAP 2.0 accumulated a lower concentration of intermediates than ASAP 1.0 (fig. S11, A and B), which indicates the effectiveness of our optimization strategies.
With the above success in ASAP 2.0, we proceeded to synthesize starch from CO2 and hydrogen by coupling the enzymatic processes with CO2 reduction by means of a previously developed inorganic catalyst, ZnO-ZrO2 (34). Because of the unfavorable conditions of CO2 hydrogenation, we developed a chemoenzymatic cascade system in ASAP 3.0 with a chemical reaction unit and an enzymatic reaction unit. To satisfy the demand of fls for a high concentration of formaldehyde and to avoid its toxicity to other enzymes (fig. S13), we further operated the enzymatic unit with two steps (Fig. 3A). In the chemical reaction unit, CO2 was chemically hydrogenated to methanol at a rate of ~0.25 g hour−1 g−1 catalyst, and the produced methanol was constantly condensed and fed into the enzymatic unit to a final concentration of ~100 mM during the first hour. In the enzymatic unit, the methanol was first converted to ~22.5 mM C3 intermediate DHA for another 1 hour by supplementing two core enzymes and auxiliary catalase (cat) and then transformed to ~1.6 g liter−1 amylose starch in the subsequent 2 hours by supplementing the remaining eight core enzymes and auxiliary components (Fig. 3A). The synthetic amylose exhibited the same deep blue color and absorption maximum as standard amylose in the presence of iodine solution (Fig. 3B).
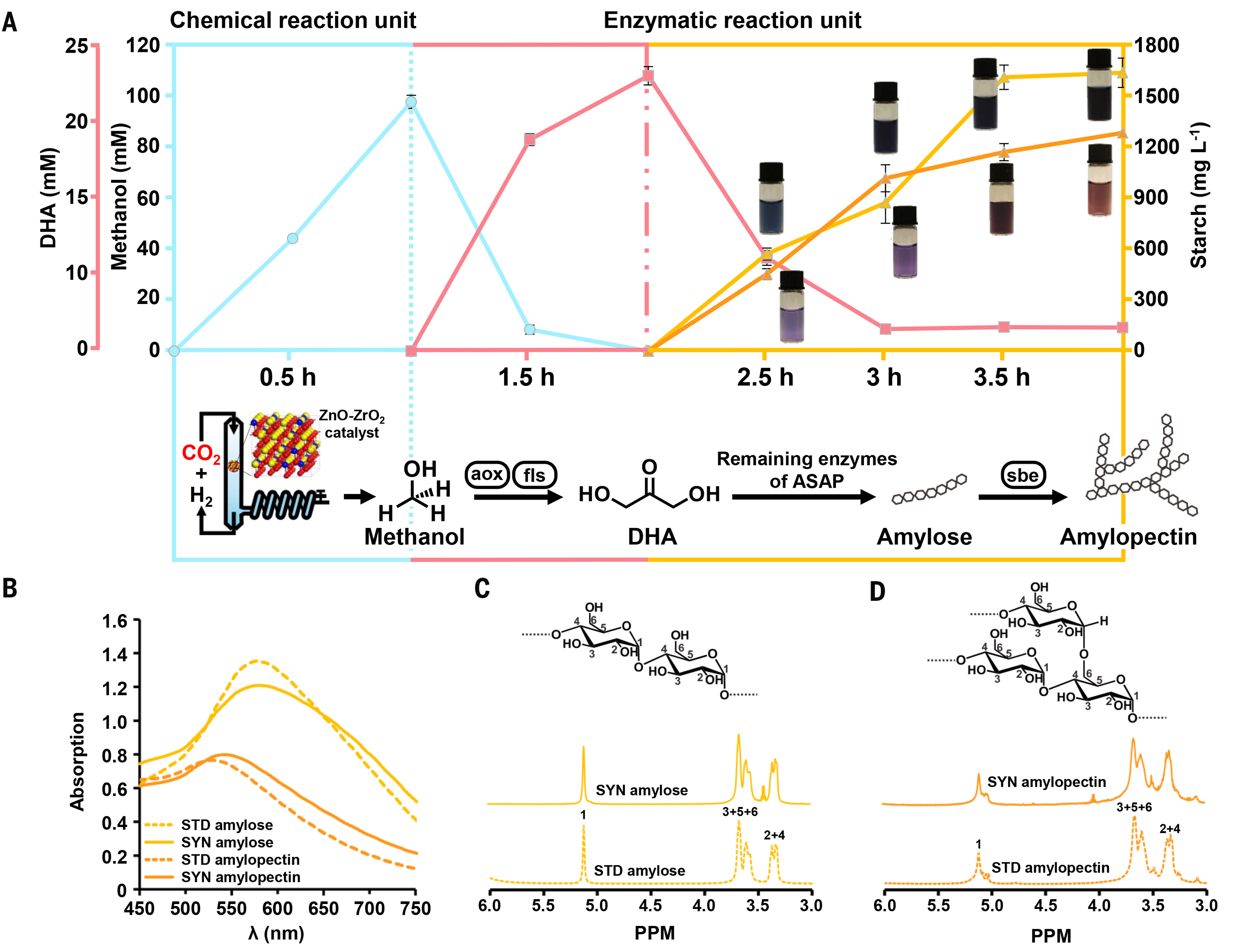
Fig. 3. Starch synthesis via ASAP from CO2.(A) (Bottom) Time course of chemoenzymatic cascade reactions for starch synthesis from CO2. sbe, starch branching enzyme. (Top) Alteration of key intermediates and starch (yellow curve indicating amylose and orange curve indicating amylopectin) during the time course. The production of amylose and amylopectin are also visualized in the vials by iodine dyeing at given time points. The reaction solution was diluted sixfold before iodine treatment. Values are means, and error bars indicate SD (n = 3 replicates). (B) Absorption spectra analysis of synthetic amylose and amylopectin after iodine treatment. The wavelength of maximum absorption (λmax) of standard (STD) and synthetic (SYN) amylose is 577 nm and λmax of standard and synthetic amylopectin is 529 and 543 nm, respectively. (C and D) 1H nuclear magnetic resonance (NMR) spectrum of standard (STD) and synthetic (SYN) amylose and amylopectin.
Natural starch contains ~20 to 30% amylose and 70 to 80% amylopectin (3). To synthesize amylopectin from CO2, we introduced a starch branching enzyme (sbe) from Vibrio vulnificus (35) in ASAP 3.1. This setup produced ~1.3 g liter−1 amylopectin within 4 hours (Fig. 3A). The synthetic amylopectin had a reddish-brown color and a comparable absorption maximum as standard amylopectin after iodine treatment (Fig. 3B). Both the synthetic amylose and amylopectin exhibited the same one to six proton nuclear magnetic resonance signals as their standard counterparts (Fig. 3, C and D).
By using spatial and temporal segregation of steps, ASAP 3.0 achieved a high starch productivity of ~410 mg liter−1 hour−1 from CO2. The starch synthesis rate of this chemoenzymatic pathway reached 22 nmol min−1 mg−1 of total catalyst and proteins, which is an 8.5-fold higher rate than that of starch synthesis via the Calvin cycle in maize (Table 1). This rate is also 5.7-fold higher than that of the synthetic crotonyl–coenzyme A (CoA)/ethylmalonyl-CoA/hydroxybutyryl-CoA (CETCH) cycle which has been recently extended into a platform to access different compounds directly from CO2 (12, 13, 36). The theoretical hydrogen-to-methanol energy efficiency (ηHME) and methanol-to-starch energy efficiency (ηMSE) of ASAP is 85 and 61%, respectively, although these values do not consider energy consumption for processes such as enzyme production and maintenance of high temperature and pressure in the chemical step (supplementary text), which will compromise the energy efficiency of ASAP in practice. With an attainable solar-to-electricity efficiency (ηSEE) of 20% (17) and electricity-to-hydrogen efficiency (ηEHE) of 85% (18) in ideal photovoltaic and water-electrolysis devices, the theoretical maximal solar-to-starch efficiency (ηSSE = ηSEE × ηEHE × ηHME × ηMSE) via ASAP will be 9%. With the estimated practical ηHME′ of 68% considering the energy for temperature and pressure in the chemical step (37), the theoretical ηSSE is adjusted to 7%, which is comparable to the theoretical photosynthetic efficiency of solar energy to biomass for C3 (4.6%) and C4 (6%) plants (38) and is 3.5 fold of the estimated theoretical solar-to-starch efficiency for plants (2%) in a natural environment (39). Cell-free, chemoenzymatic, and efficient starch synthesis from CO2 by ASAP provides an important starting point for applications such as industrial biomanufacturing of starch.
Acknowledgments
We thank P. Kang from Tianjin University for introducing us to the fundamental principles of CO2 electroreduction; Y. P. Zhang and J. Zhou (Institute of Microbiology) for granting the genome of Clostridium acetobutylicum and Syn. PCC 7942; H. Song for providing purified cat enzyme; P. Liu for molecular dynamic simulation; Z. D. Zhang and Q. C. Cao for LC-MS analysis; Y. Cai for NMR analysis; and Y. H. Yao for gas chromatography–mass spectrometry analysis. We thank G. P. Zhao, M. T. Reetz, and Z. T. Sun for advice on manuscript revision. We thank the core facility center at Tianjin Institution of Industrial Biotechnology, CAS, for instrument and technology support. We dedicate this work to the memory of Professor Arren Bar-Even, who devoted his life to synthetic pathway design and synthetic biology. Funding: This work was supported by the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Action (grant TSBICIP-KJGG-008 to T.C.), the Key Research Program of the Chinese Academy of Sciences (grant ZDRW-ZS-2016-3 to Y.M. and T.C.), the Strategic Priority Research Program of the Chinese Academy of Sciences-Precision Seed Design and Breeding (grant XDA24020103-3 to H.J.), the Tianjin Outstanding Scholar Program (to Y.M.), Youth Innovation Promotion Association of CAS (grant 2016165 to T.C.), and the National Science Fund for Excellent Young Scholars (grant 31922047 to H.J.). Author contributions: Conceptualization: Y.M. and T.C.; Methodology: Y.M., T.C., H.J., H.M., L.Z., C.L., C.Y., and Y.S.; Investigation: T.C., H.S., J.Q., L.Z., F.Z., J.Z., Z.T., X.W., J.Y., Q.Y., W.W., X.Y., H.C., and Qian Wang; Visualization: T.C., Qinhong Wang, L.Z., and H.J.; Funding acquisition: Y.M., T.C., and H.J.; Project administration: Qinhong Wang and T.C.; Supervision: Y.M.; Writing—original draft: Y.M., T.C., Qinhong Wang, H.J., and L.Z.; Writing—review and editing: Y.M., T.C., Qinhong Wang, H.J., L.Z., C.L., and Y.L. Competing interests: This work is included in patent applications by the Tianjin Institute of Industrial Biotechnology, CAS: patent 202010858974.9 covering a method of starch synthesis from C1 feedstocks and patent 202010044853.0 covering the application of the mutants of fls. Data and materials availability: All data are available in the main text or the supplementary materials.
References and Notes
1 - P. L. Keeling, A. M. Myers, Biochemistry and genetics of starch synthesis. Annu. Rev. Food Sci. Technol. 1, 271–303 (2010).
2 - R. Höfer, “Sugar- and starch-based biorefineries” in Industrial Biorefineries and White Biotechnology, A. Pandey, R. Höfer, M. Taherzadeh, M. Nampoothiri, C. Larroche, P. L. Keeling, A. M. Myers, Eds. (Elsevier, 2015), chap. 4A, pp. 157–235.
3 - R. L. Whistler, J. R. Daniel, “Molecular structure of starch” in Starch: Chemistry and Technology, R. L. Whistler, J. N. Bemiller, E. F. Paschall, Eds. (Academic Press, ed. 2, 1984), pp. 153–182.
4 - T. Saithong, A. Meechai, S. Cheevadhanarak, S. Bhumiratana, A formal path inference of starch biosynthesis via mathematical modelling of metabolic changes in excess CO2. J. Comput. Sci. Syst. Biol. 5, 24–37 (2012).
5 - M. R. Abt, S. C. Zeeman, Evolutionary innovations in starch metabolism. Curr. Opin. Plant Biol. 55, 109–117 (2020).
6 - J. Li, E. Baroja-Fernández, A. Bahaji, F. J. Muñoz, M. Ovecka, M. Montero, M. T. Sesma, N. Alonso-Casajús, G. Almagro, A. M. Sánchez-López, M. Hidalgo, M. Zamarbide, J. Pozueta-Romero, Enhancing sucrose synthase activity results in increased levels of starch and ADP-glucose in maize (Zea mays L.) seed endosperms. Plant Cell Physiol. 54, 282–294 (2013).
7 - N. Li, S. Zhang, Y. Zhao, B. Li, J. Zhang, Over-expression of AGPase genes enhances seed weight and starch content in transgenic maize. Planta 233, 241–250 (2011).
8 - M. Hakata, M. Kuroda, T. Miyashita, T. Yamaguchi, M. Kojima, H. Sakakibara, T. Mitsui, H. Yamakawa, Suppression of α-amylase genes improves quality of rice grain ripened under high temperature. Plant Biotechnol. J. 10, 1110–1117 (2012).
9 - A. Bahaji, J. Li, A. M. Sánchez-López, E. Baroja-Fernández, F. J. Muñoz, M. Ovecka, G. Almagro, M. Montero, I. Ezquer, E. Etxeberria, J. Pozueta-Romero, Starch biosynthesis, its regulation and biotechnological approaches to improve crop yields. Biotechnol. Adv. 32, 87–106 (2014).
10 - A. Bar-Even, E. Noor, N. E. Lewis, R. Milo, Design and analysis of synthetic carbon fixation pathways. Proc. Natl. Acad. Sci. U.S.A. 107, 8889–8894 (2010).
11 - A. Satanowski, B. Dronsella, E. Noor, B. Vögeli, H. He, P. Wichmann, T. J. Erb, S. N. Lindner, A. Bar-Even, Awakening a latent carbon fixation cycle in Escherichia coli. Nat. Commun. 11, 5812 (2020).
12 - M. Scheffen, D. G. Marchal, T. Beneyton, S. K. Schuller, M. Klose, C. Diehl, J. Lehmann, P. Pfister, M. Carrillo, H. He, S. Aslan, N. S. Cortina, P. Claus, D. Bollschweiler, J.-C. Baret, J. M. Schuller, J. Zarzycki, A. Bar-Even, T. J. Erb, A new-to-nature carboxylation module to improve natural and synthetic CO2 fixation. Nat. Catal. 4, 105–115 (2021).
13 - T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina, T. J. Erb, A synthetic pathway for the fixation of carbon dioxide in vitro. Science 354, 900–904 (2016).
14 - T. E. Miller, T. Beneyton, T. Schwander, C. Diehl, M. Girault, R. McLean, T. Chotel, P. Claus, N. S. Cortina, J.-C. Baret, T. J. Erb, Light-powered CO2 fixation in a chloroplast mimic with natural and synthetic parts. Science 368, 649–654 (2020).
15 - T. P. Korman, P. H. Opgenorth, J. U. Bowie, A synthetic biochemistry platform for cell free production of monoterpenes from glucose. Nat. Commun. 8, 15526 (2017).
16 - S. Sherkhanov, T. P. Korman, S. Chan, S. Faham, H. Liu, M. R. Sawaya, W. T. Hsu, E. Vikram, T. Cheng, J. U. Bowie, Isobutanol production freed from biological limits using synthetic biochemistry. Nat. Commun. 11, 4292 (2020).
17 - M. Green, E. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis, X. J. Hao, Solar cell efficiency tables (version 57). Prog. Photovolt. Res. Appl. 29, 3–15 (2021).
18 - S. Shiva Kumar, V. Himabindu, Hydrogen production by PEM water electrolysis – A review. Mater. Sci. Energy Technol. 2, 442–454 (2019).
19 - Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo, M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
20 - R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan, Y.-G. Yao, CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 10, 5698 (2019).
21 - J. B. Siegel, A. L. Smith, S. Poust, A. J. Wargacki, A. Bar-Even, C. Louw, B. W. Shen, C. B. Eiben, H. M. Tran, E. Noor, J. L. Gallaher, J. Bale, Y. Yoshikuni, M. H. Gelb, J. D. Keasling, B. L. Stoddard, M. E. Lidstrom, D. Baker, Computational protein design enables a novel one-carbon assimilation pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 3704–3709 (2015).
22 - R. Caspi, R. Billington, L. Ferrer, H. Foerster, C. A. Fulcher, I. M. Keseler, A. Kothari, M. Krummenacker, M. Latendresse, L. A. Mueller, Q. Ong, S. Paley, P. Subhraveti, D. S. Weaver, P. D. Karp, The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44, D471–D480 (2016).
23 - N. Hadadi, J. Hafner, A. Shajkofci, A. Zisaki, V. Hatzimanikatis, ATLAS of biochemistry: A repository of all possible biochemical reactions for synthetic biology and metabolic engineering studies. ACS Synth. Biol. 5, 1155–1166 (2016).
24 - X. Yang, Q. Yuan, H. Luo, F. Li, Y. Mao, X. Zhao, J. Du, P. Li, X. Ju, Y. Zheng, Y. Chen, Y. Liu, H. Jiang, Y. Yao, H. Ma, Y. Ma, Systematic design and in vitro validation of novel one-carbon assimilation pathways. Metab. Eng. 56, 142–153 (2019).
25 - A. Ebrahim, J. A. Lerman, B. O. Palsson, D. R. Hyduke, COBRApy: Constraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7, 74 (2013).
26 - T. J. Erb, P. R. Jones, A. Bar-Even, Synthetic metabolism: Metabolic engineering meets enzyme design. Curr. Opin. Chem. Biol. 37, 56–62 (2017).
27 - A. Chou, J. M. Clomburg, S. Qian, R. Gonzalez, 2-Hydroxyacyl-CoA lyase catalyzes acyloin condensation for one-carbon bioconversion. Nat. Chem. Biol. 15, 900–906 (2019).
28 - Y. S. Tai, K. Zhang, C1 metabolism redesigned. Nat. Chem. Biol. 11, 384–386 (2015).
29 - S. Poust, J. Piety, A. Bar-Even, C. Louw, D. Baker, J. D. Keasling, J. B. Siegel, Mechanistic analysis of an engineered enzyme that catalyzes the formose reaction. ChemBioChem 16, 1950–1954 (2015).
30 - J. K. Hines, C. E. Kruesel, H. J. Fromm, R. B. Honzatko, Structure of inhibited fructose-1,6-bisphosphatase from Escherichia coli: Distinct allosteric inhibition sites for AMP and glucose 6-phosphate and the characterization of a gluconeogenic switch. J. Biol. Chem. 282, 24697–24706 (2007).
31 - J. S. Yang, S. W. Seo, S. Jang, G. Y. Jung, S. Kim, Rational engineering of enzyme allosteric regulation through sequence evolution analysis. PLOS Comput. Biol. 8, e1002612 (2012).
32 - C. R. Meyer, J. A. Bork, S. Nadler, J. Yirsa, J. Preiss, Site-directed mutagenesis of a regulatory site of Escherichia coli ADP-glucose pyrophosphorylase: The role of residue 336 in allosteric behavior. Arch. Biochem. Biophys. 353, 152–159 (1998).
33 - C. R. Meyer, J. Yirsa, B. Gott, J. Preiss, A kinetic study of site-directed mutants of Escherichia coli ADP-glucose pyrophosphorylase: The role of residue 295 in allosteric regulation. Arch. Biochem. Biophys. 352, 247–254 (1998).
34 - J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu, C. Li, A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 3, e1701290 (2017).
35 - H. J. Jo, S. Park, H. G. Jeong, J. W. Kim, J. T. Park, Vibrio vulnificus glycogen branching enzyme preferentially transfers very short chains: N1 domain determines the chain length transferred. FEBS Lett. 589, 1089–1094 (2015).
36 - S. Sundaram, C. Diehl, N. S. Cortina, J. Bamberger, N. Paczia, T. J. Erb, A modular in vitro platform for the production of terpenes and polyketides from CO2. Angew. Chem. Int. Ed. 60, 16420–16425 (2021).
37 - E. S. Van-Dal, C. Bouallou, Design and simulation of a methanol production plant from CO2 hydrogenation. J. Clean. Prod. 57, 38–45 (2013).
38 - X. G. Zhu, S. P. Long, D. R. Ort, What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Curr. Opin. Biotechnol. 19, 153–159 (2008).
39 - Y. H. P. Zhang, C. You, H. Chen, R. Feng, “Surpassing photosynthesis: high-efficiency and scalable CO2 utilization through artificial photosynthesis” in Recent Advances in Post-Combustion CO2 Capture Chemistry. M. Attalla, Ed. (American Chemical Society, 2012), pp. 275–292.
40 - C. S. Kuehn, J. G. Linn, D. G. Johnson, H. G. Jung, M. I. Endres, Effect of feeding silages from corn hybrids selected for leafiness or grain to lactating dairy cattle. J. Dairy Sci. 82, 2746–2755 (1999).
41 - M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
42 - P. A. Lanzetta, L. J. Alvarez, P. S. Reinach, O. A. Candia, An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97 (1979).
43 - M. A. Tang, H. Motoshima, K. Watanabe, Cold adaptation: Structural and functional characterizations of psychrophilic and mesophilic acetate kinase. Protein J. 33, 313–322 (2014).
44 - H. Bi, Y. Bai, T. Cai, Y. Zhuang, X. Liang, X. Zhang, T. Liu, Y. Ma, Engineered short branched-chain acyl-CoA synthesis in E. coli and acylation of chloramphenicol to branched-chain derivatives. Appl. Microbiol. Biotechnol. 97, 10339–10348 (2013).
45 - M. Molin, J. Norbeck, A. Blomberg, Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J. Biol. Chem. 278, 1415–1423 (2003).
46 - E. González-Mondragón, R. A. Zubillaga, E. Saavedra, M. E. Chánez-Cárdenas, R. Pérez-Montfort, A. Hernández-Arana, Conserved cysteine 126 in triosephosphate isomerase is required not for enzymatic activity but for proper folding and stability. Biochemistry 43, 3255–3263 (2004).
47 - O. N. Rozova, V. N. Khmelenina, I. I. Mustakhimov, A. S. Reshetnikov, Y. A. Trotsenko, Characterization of recombinant fructose-1,6-bisphosphate aldolase from Methylococcus capsulatus Bath. Biochemistry (Mosc.) 75, 892–898 (2010).
48 - C. V. Iancu, S. Mukund, H. J. Fromm, R. B. Honzatko, R-state AMP complex reveals initial steps of the quaternary transition of fructose-1,6-bisphosphatase. J. Biol. Chem. 280, 19737–19745 (2005).
49 - S. Schneider, T. Sandalova, G. Schneider, G. A. Sprenger, A. K. Samland, Replacement of a phenylalanine by a tyrosine in the active site confers fructose-6-phosphate aldolase activity to the transaldolase of Escherichia coli and human origin. J. Biol. Chem. 283, 30064–30072 (2008).
50 - D. Mathur, L. C. Garg, Functional phosphoglucose isomerase from Mycobacterium tuberculosis H37Rv: Rapid purification with high yield and purity. Protein Expr. Purif. 52, 373–378 (2007).
51 - C. You, S. Myung, Y. H. Zhang, Facilitated substrate channeling in a self-assembled trifunctional enzyme complex. Angew. Chem. Int. Ed. 51, 8787–8790 (2012).
52 - X. Ji, Z. Su, P. Wang, G. Ma, S. Zhang, Tethering of nicotinamide adenine dinucleotide inside hollow nanofibers for high-yield synthesis of methanol from carbon dioxide catalyzed by coencapsulated multienzymes. ACS Nano 9, 4600–4610 (2015).
53 - S. Fregert, I. Dahlquist, B. Gruvberger, A simple method for the detection of formaldehyde. Contact Dermatitis 10, 132–134 (1984).
54 - G. Hernández-Alcántara, G. Garza-Ramos, G. M. Hernández, A. Gómez-Puyou, R. Pérez-Montfort, Catalysis and stability of triosephosphate isomerase from Trypanosoma brucei with different residues at position 14 of the dimer interface. Characterization of a catalytically competent monomeric enzyme. Biochemistry 41, 4230–4238 (2002).
55 - C. A. Smith, T. Kortemme, Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 380, 742–756 (2008).
56 - J. D. Durrant, L. Votapka, J. Sørensen, R. E. Amaro, POVME 2.0: An enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput. 10, 5047–5056 (2014).
57 - I. W. Davis, D. Baker, RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 385, 381–392 (2009).
58 - S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang, S. H. Bryant, PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–D1213 (2016).
59 - N. M. O’Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch, G. R. Hutchison, Open Babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011).
60 - S. C. Li, Y. K. Ng, Calibur: A tool for clustering large numbers of protein decoys. BMC Bioinformatics 11, 25 (2010).
61 - E. Lilkova, P. Petkov, N. Ilieva, and L. Litov, The PyMOL Molecular Graphics System Version 2.0. (Software, Schrödinger LLC., 2015).
62 - C. You, H. Chen, S. Myung, N. Sathitsuksanoh, H. Ma, X. Z. Zhang, J. Li, Y. H. Zhang, Enzymatic transformation of nonfood biomass to starch. Proc. Natl. Acad. Sci. U.S.A. 110, 7182–7187 (2013).
63 - P. Kang, S. Zhang, T. J. Meyer, M. Brookhart, Rapid selective electrocatalytic reduction of carbon dioxide to formate by an iridium pincer catalyst immobilized on carbon nanotube electrodes. Angew. Chem. Int. Ed. 53, 8709–8713 (2014).
64 - R. K. Singh, R. Singh, D. Sivakumar, S. Kondaveeti, T. Kim, J. Li, B. H. Sung, B.-K. Cho, D. R. Kim, S. C. Kim, V. C. Kalia, Y.-H. P. J. Zhang, H. Zhao, Y. C. Kang, J.-K. Lee, Insights into cell-free conversion of CO2 to chemicals by a multienzyme cascade reaction. ACS Catal. 8, 11085–11093 (2018).
65 - R. F. Say, G. Fuchs, Fructose 1,6-bisphosphate aldolase/phosphatase may be an ancestral gluconeogenic enzyme. Nature 464, 1077–1081 (2010).
66 - S. Fushinobu, H. Nishimasu, D. Hattori, H. J. Song, T. Wakagi, Structural basis for the bifunctionality of fructose-1,6-bisphosphate aldolase/phosphatase. Nature 478, 538–541 (2011).
67 - A. M. Smith, S. C. Zeeman, S. M. Smith, Starch degradation. Annu. Rev. Plant Biol. 56, 73–98 (2005).
68 - K. Ohdan, K. Fujii, M. Yanase, T. Takaha, T. Kuriki, Enzymatic synthesis of amylose. Biocatal. Biotransform. 24, 77–81 (2006).
69 - J. I. Kadokawa, Enzymatic synthesis of functional amylosic materials and amylose analog polysaccharides. Methods Enzymol. 627, 189–213 (2019).
70 - F. Y. H. Chen, H. W. Jung, C. Y. Tsuei, J. C. Liao, Converting Escherichia coli to a synthetic methylotroph growing solely on methanol. Cell 182, 933–946.e14 (2020).
71 - J. Ren, P. Yao, S. Yu, W. Dong, Q. Chen, J. Feng, Q. Wu, D. Zhu, An unprecedented effective enzymatic carboxylation of phenols. ACS Catal. 6, 564–567 (2016).
72 - A. Chang, I. Schomburg, S. Placzek, L. Jeske, M. Ulbrich, M. Xiao, C. W. Sensen, D. Schomburg, BRENDA in 2015: Exciting developments in its 25th year of existence. Nucleic Acids Res. 43, D439–D446 (2015).
73 - X. Garrabou, J. A. Castillo, C. Guérard-Hélaine, T. Parella, J. Joglar, M. Lemaire, P. Clapés, Asymmetric self- and cross-aldol reactions of glycolaldehyde catalyzed by D-fructose-6-phosphate aldolase. Angew. Chem. Int. Ed. 48, 5521–5525 (2009).
74 - V. Helaine, R. Mahdi, G. V. Sudhir Babu, V. de Berardinis, R. Wohlgemuth, M. Lemaire, C. Guerard-Helaine, Straightforward synthesis of terminally phosphorylated l-sugars via multienzymatic cascade reactions. Adv. Synth. Catal. 357, 1703–1708 (2015).
75 - A. Szekrenyi, X. Garrabou, T. Parella, J. Joglar, J. Bujons, P. Clapés, Asymmetric assembly of aldose carbohydrates from formaldehyde and glycolaldehyde by tandem biocatalytic aldol reactions. Nat. Chem. 7, 724–729 (2015).
76 - A. Flamholz, E. Noor, A. Bar-Even, R. Milo, eQuilibrator—The biochemical thermodynamics calculator. Nucleic Acids Res. 40, D770–D775 (2012).
77 - A. Krog, T. M. B. Heggeset, J. E. N. Müller, C. E. Kupper, O. Schneider, J. A. Vorholt, T. E. Ellingsen, T. Brautaset, Methylotrophic Bacillus methanolicus encodes two chromosomal and one plasmid born NAD+ dependent methanol dehydrogenase paralogs with different catalytic and biochemical properties. PLOS ONE 8, e59188 (2013).
78 - X. Lu, Y. Liu, Y. Yang, S. Wang, Q. Wang, X. Wang, Z. Yan, J. Cheng, C. Liu, X. Yang, H. Luo, S. Yang, J. Gou, L. Ye, L. Lu, Z. Zhang, Y. Guo, Y. Nie, J. Lin, S. Li, C. Tian, T. Cai, B. Zhuo, H. Ma, W. Wang, Y. Ma, Y. Liu, Y. Li, H. Jiang, Constructing a synthetic pathway for acetyl-coenzyme A from one-carbon through enzyme design. Nat. Commun. 10, 1378 (2019).
79 - T. Senoura, A. Asao, Y. Takashima, N. Isono, S. Hamada, H. Ito, H. Matsui, Enzymatic characterization of starch synthase III from kidney bean (Phaseolus vulgaris L.). FEBS J. 274, 4550–4560 (2007).
80 - H. Choe, J. C. Joo, D. H. Cho, M. H. Kim, S. H. Lee, K. D. Jung, Y. H. Kim, Efficient CO2-reducing activity of NAD-dependent formate dehydrogenase from Thiobacillus sp. KNK65MA for formate production from CO2 gas. PLOS ONE 9, e103111 (2014).
81 - H. Zheng, Z. Yu, W. Shu, X. Fu, X. Zhao, S. Yang, M. Tan, J. Xu, Y. Liu, H. Song, Ethanol effects on the overexpression of heterologous catalase in Escherichia coli BL21 (DE3). Appl. Microbiol. Biotechnol. 103, 1441–1453 (2019).
82 - K. T. Shum, E. L. H. Lui, S. C. K. Wong, P. Yeung, L. Sam, Y. Wang, R. M. Watt, J. A. Tanner, Aptamer-mediated inhibition of Mycobacterium tuberculosis polyphosphate kinase 2. Biochemistry 50, 3261–3271 (2011).
83 - S. Avaeva, P. Ignatov, S. Kurilova, T. Nazarova, E. Rodina, N. Vorobyeva, V. Oganessyan, E. Harutyunyan, Escherichia coli inorganic pyrophosphatase: Site-directed mutagenesis of the metal binding sites. FEBS Lett. 399, 99–102 (1996).